In this tutorial, the main features of DAISIEprep are explained. DAISIEprep (Lambert et al.) is an R package that facilitates formatting of data for subsequent use in DAISIE.
![]()
A typical DAISIEprep pipeline is as follows:
- Load phylogenetic tree and island distribution data (endemic, nonendemic to the island) into R.
- Assign an island endemicity status (endemic, nonendemic, not present) to each of the species on the tree.
- Automatically extract times of colonisation of the island and diversification within the island from the phylogenies.
- Add any missing species.
- Format data for DAISIE.
The tutorial is divided into 3 sections:
Single phylogeny example - Learn how to extract and format island data for running DAISIE based on a single phylogenetic tree.
Multiple phylogenies example - Learn how to extract and format island data for running DAISIE based for cases where you have multiple phylogenetic trees (e.g. different phylogenies from different literature sources including different island lineages).
Adding missing species - How to add missing species, lineages, etc, to your DAISIE data list.
Load the required packages:
1. Single Phylogeny Example
In this section we demonstrate a simple example of extracting and formatting data from a single phylogeny. We use an example phylogeny of an imaginary clade of plant species, which includes 2 species occurring on an example focal island.
Load the phylogeny
First we load our example phylogeny (file included in the
DAISIEprep R package, that is why we use this complicated
code:
data("plant_phylo")For your own trees you would normally use something like
read.tree(), read.nexus(), load()
specifying the path to your tree file or R phylo object).
Object plant_phylo we have just loaded is a “typical”
phylogeny of class ‘phylo’.
class(plant_phylo)
#> [1] "phylo"Let’s plot the phylogeny in order to visualise it. As you can see, it has different species of plants.
plot(plant_phylo, underscore = TRUE)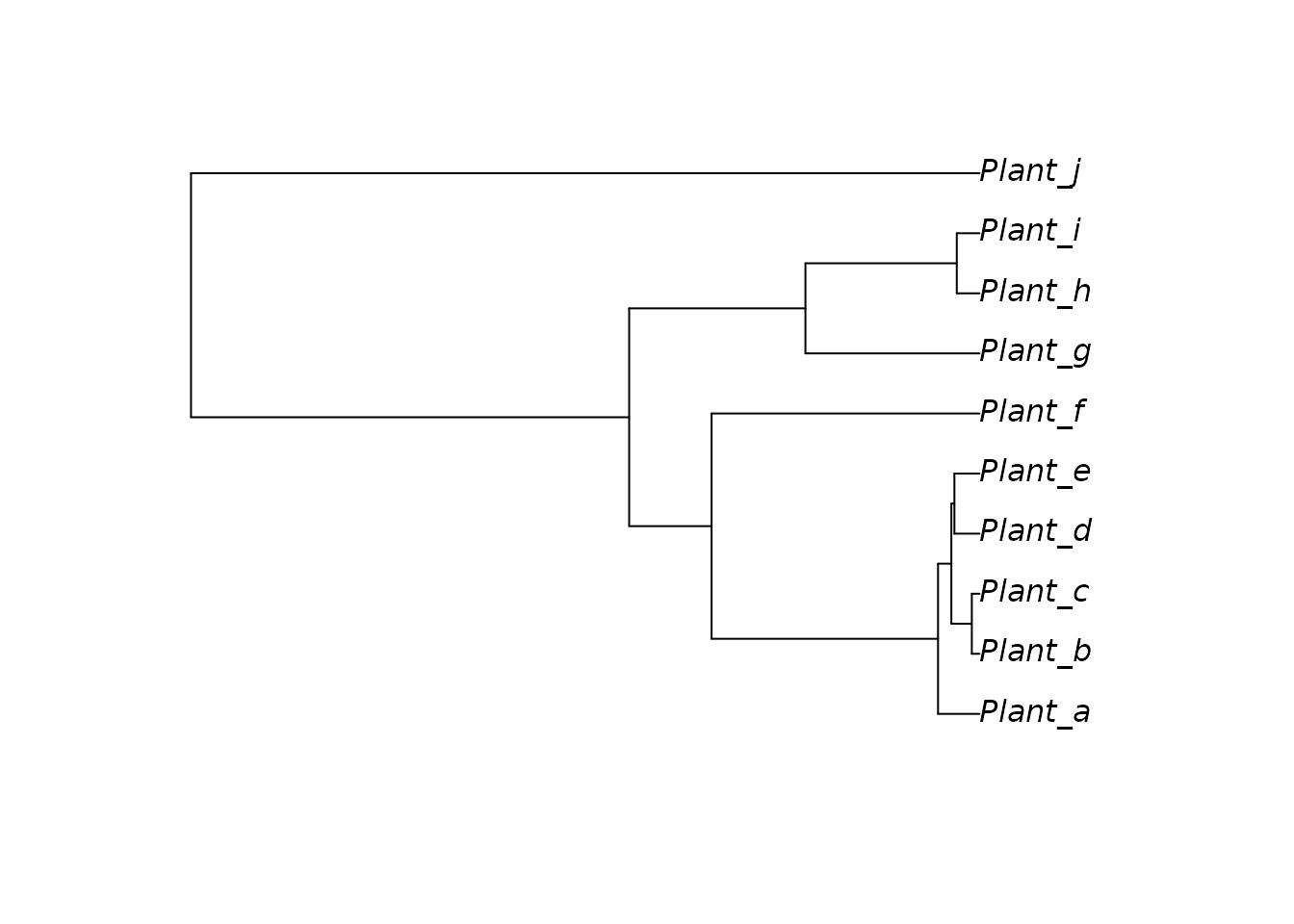
Important: DAISIEprep requires the tip labels
(taxon names) in the phylogeny to be formatted as genus name and species
name separated by an underscore (e.g. “Canis_lupus” or
“Plant_a”). They can also optionally have tags appended after
the species name (separated by underscore, e.g. “Canis_lupus_123”;
“Plant_a_123”; “Canis_lupus_familiaris_123”), which is common if there
are multiple tips in the phylogeny for a single species, e.g. when
multiple populations or multiple subspecies have been sampled. Samples
with the same Genus_species name on the tip label will be considered to
be of the same species, even if they have subsequent
sampling or subspecies tags.
So in the example above we have turned on the option
underscore = TRUE in the plotting function, so that you can
see that there is an underscore between the words on the tip names. In
this case, “Plant” can be viewed as the genus epithet in a binomial
name, and the letters after the underscore as the species epithet (for
instance “Genus_speciesA” or in this case “Plant_a”).
Currently DAISIEprep does not deal well with island species that are not monophyletic, because it does not know how to make a decision on how many colonisation events to infer. This needs some expert input from the user (e.g. you may consider that some tips are incorrectly placed, or perhaps the species really is polyphyletic). So we recommend you make decisions for these cases before running the package.
Convert the phylogeny to phylo4
The next step is to convert the phylogeny into a phylo4
class, defined in the package phylobase. This allows users
to easily work with data for each tip in the phylogeny, for example
whether they are endemic to the island or not.
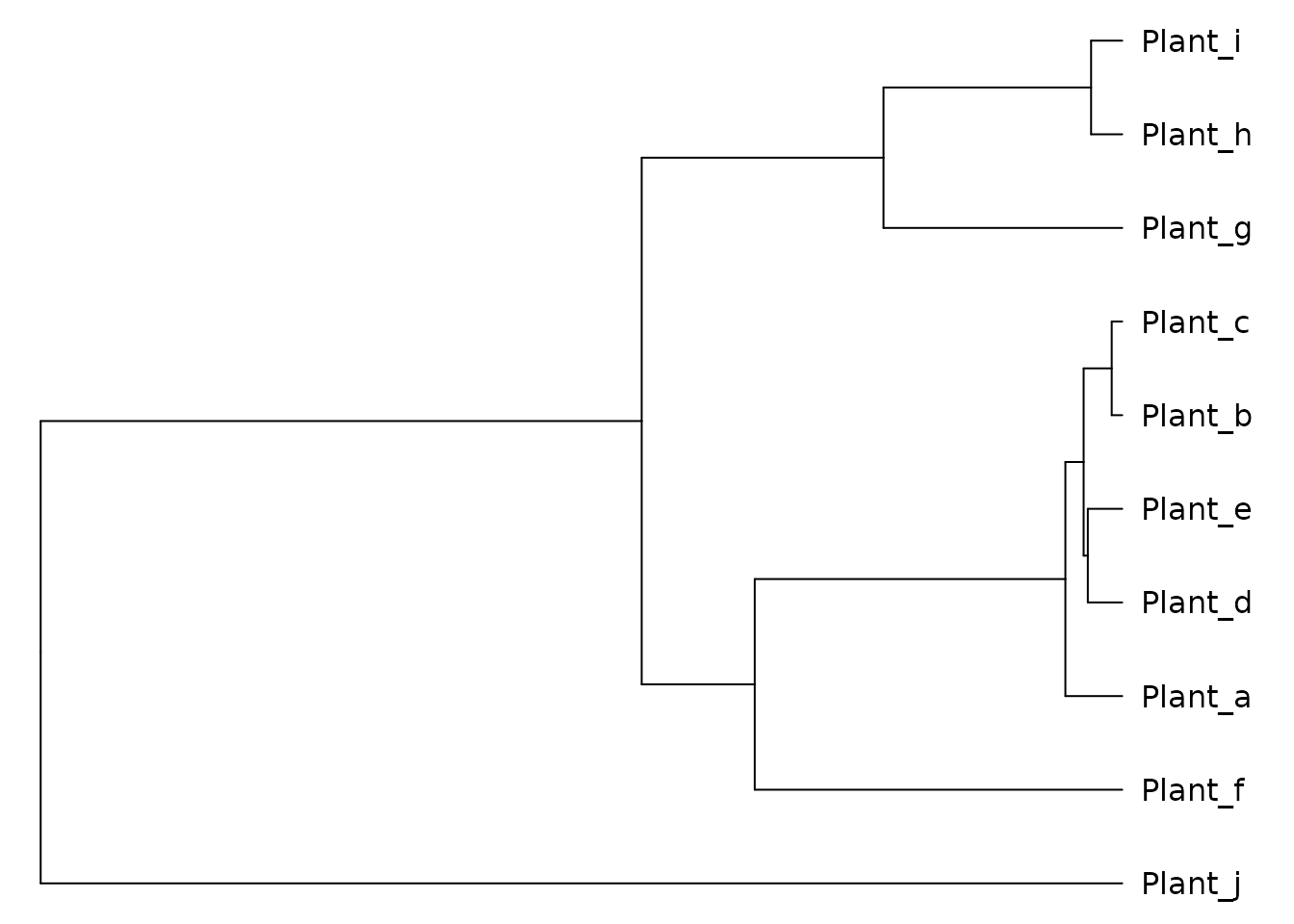
Now we have a phylogeny in the phylo4 format to which we
can easily append data.
Add endemicity statuses
We now need to add to the phylogeny information about which species are present on the island and what their endemicity statuses are. Species that occur on the island can either be “endemic” (occur only on the island), or “nonendemic” (occur on the island but also on another landmass).
In the example plant phylogeny we are running here, only two plant species in the phylogeny occur on the island. One of them, “Plant_i”, is endemic to the island. The other, “Plant_g”, is a nonendemic species. Let’s create a data frame linking each island species to their endemicity status. (For your own data, you can create this table in a separate editor and then load it into R as a data.frame).
We recommend using the true endemicity status (“endemic” or “nonendemic”) for all cases. However, there is an exception: cases where one or more species from an endemic island radiation have colonised the mainland. These species are embedded in the island radiation, have evolved on the island, and later expanded their range to the mainland. Technically these species are nonendemic because they are found on both the island and mainland. However, from the perspective of the DAISIE model, we recommend scoring such species as endemic. This is because scoring them as nonendemic may lead to complex biogeographical reconstruction that may end up breaking up an endemic island radiation into multiple lineages; and also because this may lead to an underestimation of the speciation rates on the island. By classifying those species as endemic we can avoid these problems.
island_species <- data.frame(
tip_labels = c("Plant_i", "Plant_g"),
tip_endemicity_status = c("endemic", "nonendemic")
)
island_species
#> tip_labels tip_endemicity_status
#> 1 Plant_i endemic
#> 2 Plant_g nonendemicThe island_species data frame produced above specifies
the island endemicity status of only the species that are found on the
island. We can generate the rest of the endemicity statuses for those
species that are in the phylogeny but are not present on the island
using create_endemicity_status(). All such species will be
assigned “not_present”.
endemicity_status <- create_endemicity_status(
phylo = plant_phylo,
island_species = island_species
)Next, we can add the endemicity data to our phylogenetic tree using
features of phylo4d class, again from the
phylobase package. This call is designed for phylogenetic
and trait data to be stored together. The endemicity status needs to be
in a data frame format in order for this to work correctly.
phylod <- phylobase::phylo4d(plant_phylo, endemicity_status)We can now visualise our phylogeny with the island endemicity
statuses plotted at the tips. This uses the ggtree and
ggplot2 packages.
plot_phylod(phylod = phylod)
Create island_tbl object
Now that we know the tips in the phylogeny that are present on the
island, we can extract their colonisation and branching times, to form
our island community data set that can be used in the
DAISIE R package to fit likelihood models of island
colonisation and diversification. Before we extract species, we will
first create an object to store all of the island colonists’
information. This uses the island_tbl class introduced in
this package (DAISIEprep). This island_tbl
object can then easily be converted to a DAISIE data list using the
function create_daisie_data (more information on this
below).
island_tbl <- island_tbl()
island_tbl
#> Class: Island_tbl
#> [1] clade_name status missing_species col_time
#> [5] col_max_age branching_times min_age species
#> [9] clade_type
#> <0 rows> (or 0-length row.names)We can see that this is an object containing an empty data frame. In order to fill this data frame with information on the island colonisation and diversification events we can run the steps below.
Extract data with the DAISIEprep “min” algorithm
The function extract_island_species() is the main
function in DAISIEprep to extract data from a phylogeny. In
the example below, we use the “min” extraction algorithm. The “min”
algorithm extracts island community data with the assumptions of the
DAISIE model (i.e. no back-colonisation from the island to the
mainland).
island_tbl <- extract_island_species(
phylod = phylod,
extraction_method = "min"
)
island_tbl
#> Class: Island_tbl
#> clade_name status missing_species col_time col_max_age branching_times
#> 1 Plant_g nonendemic 0 0.38003405 FALSE NA
#> 2 Plant_i endemic 0 0.04960523 FALSE NA
#> min_age species clade_type
#> 1 NA Plant_g 1
#> 2 NA Plant_i 1Each row in the island_tbl corresponds to a separate
colonisation of the island. In this case, two colonist lineages
were identified using the ‘min’ extraction algorithm, one endemic and
another nonendemic.
Extract data with the DAISIEprep “asr” algorithm
The “min” algorithm sometimes does a good job, but we recommend using the “asr” algorithm instead when back-colonisation events are present in the data (for example, one species within a large endemic island radiation colonised another island or mainland). To use the “asr” algorithm to extract the most likely colonisations inferred in an ancestral state reconstruction, we need to first estimate the probability of the ancestors of the island species being present on the island, in order to determine the time of colonisation. To do this, we can fit one of many ancestral state reconstruction methods (see the “Extending_asr” tutorial). Here we use maximum parsimony as it is a simple method for reconstructing ancestral species areas (i.e. present on the island, or not present on the island). First, we translate our extant species endemicity status to a numeric representation of whether that species is on the island, using the following function:
phylod <- add_asr_node_states(phylod = phylod, asr_method = "parsimony")Now we can plot the phylogeny, which this time includes the node labels for the presence/absence on the island in ancestral nodes.
plot_phylod(phylod = phylod)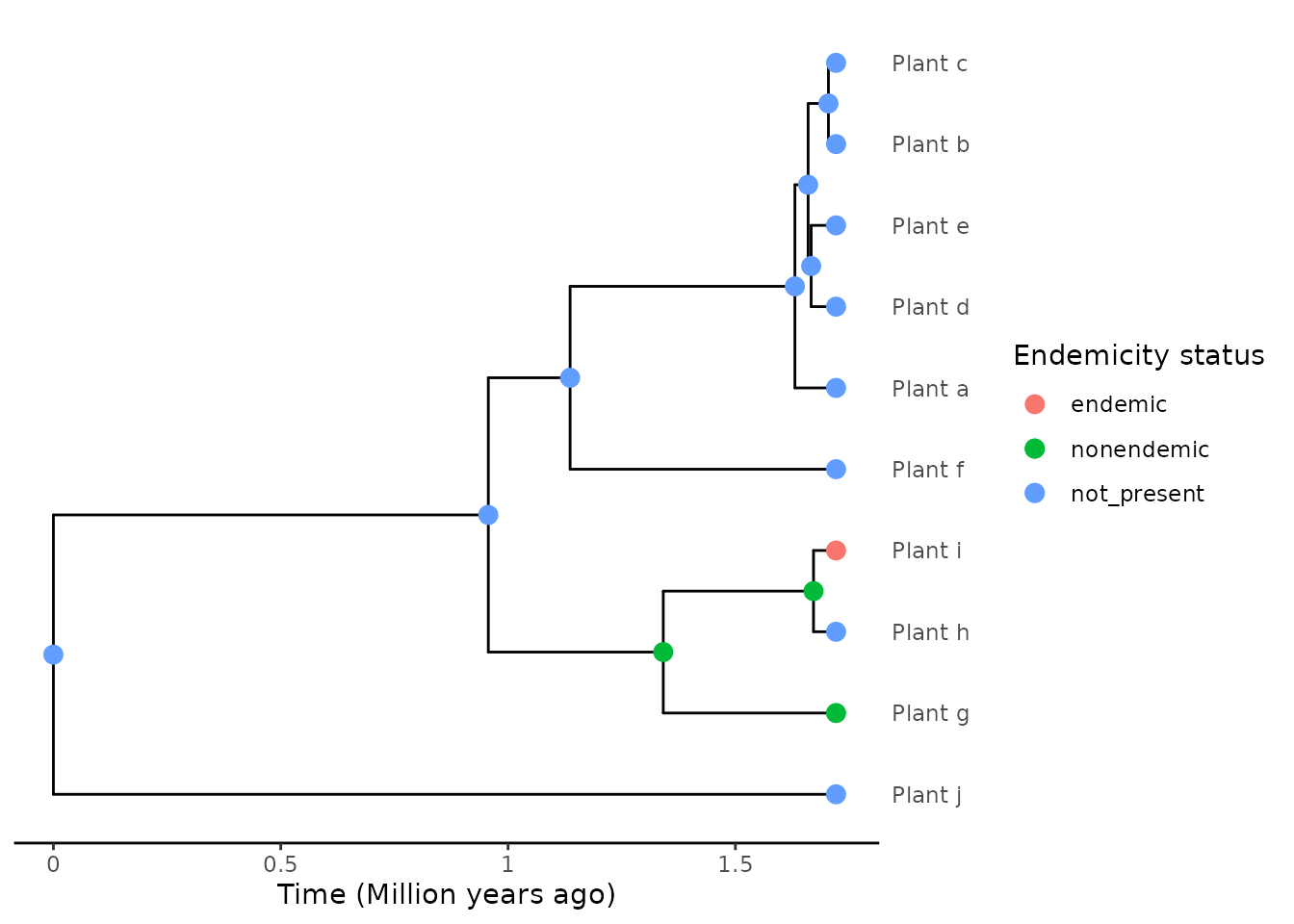
Sidenote: if you are wondering what the probabilities are at each
node and whether this should influence your decision to pick a
preference for island or mainland when the likelihoods for each state
are equal, we can plot the probabilities at the nodes to visualise the
ancestral state reconstruction using
plot_phylod(phylod = phylod, node_pies = TRUE).
Now we can extract island colonisation and diversification times from the phylogeny using the reconstructed ancestral states of island presence/absence.
island_tbl <- extract_island_species(
phylod = phylod,
extraction_method = "asr"
)If you are sure that all species you have scored as
“nonendemic” are the result of separate colonisation events (e.g., when
scoring the endemicity statuses you classified nonendemic species that
are embedded in an island radiation as endemic), then we recommend
setting force_nonendemic_singleton = TRUE. This will ensure
that all non-endemic species are extracted as separate lineages (avoids
groups of closely related non-endemic species being erroneously lumped
into an island clade due to the reconstruction).
island_tbl
#> Class: Island_tbl
#> clade_name status missing_species col_time col_max_age branching_times
#> 1 Plant_g endemic 0 0.7648553 FALSE 0.380034....
#> min_age species clade_type
#> 1 NA Plant_g,.... 1As you can see, in this case using the “asr” algorithm we
find a single colonisation of the island, as can be seen by the
fact that the island_tbl only has one row. The two island
species are inferred to result from a single colonisation of the island,
and the non-island species within that clade (“Plant_h”) is inferred to
have resulted from a back-colonisation from the island to the
mainland.
Format data to prepare if for DAISIE
Now that we have the island_tbl we can convert this to
the DAISIE data list to be used by the DAISIE inference model.
To convert to the DAISIE data list (i.e. the input data of the DAISIE
inference model) we use create_daisie_data(), providing the
island_tbl as input. We also need to specify:
- The age of the island or archipelago. Here we use an island age of
one million years (
island_age = 1). - Whether the colonisation times extracted from the phylogenetic data
should be considered precise (
precise_col_time = TRUE). We will not discuss the details of this here, but briefly by setting this toTRUEthis will tell the DAISIE model that the colonisation times are known. Settingprecise_col_time = FALSEwill change tell the DAISIE model that the colonisation time is uncertain and should interpret this as the upper limit to the time of colonisation and integrate over the uncertainty between this point and either the present time or to the first branching point (either speciation or divergence into subspecies). - The number of species in the mainland source pool. Here we set it to
100 (
num_mainland_species = 100). This will be used to calculate the number of species that could have potentially colonised the island but have not. When we refer to the mainland pool, this does not necessarily have to be a continent, it could be a different island if the source of species immigrating to an island are largely from another nearby island (a possible example of this could be Madagascar being the source of species colonising Comoros). This information is used by the DAISIE model to calculate the colonisation rate of the island.
data_list <- create_daisie_data(
data = island_tbl,
island_age = 1,
num_mainland_species = 100,
precise_col_time = TRUE
)Below we show two elements of the DAISIE data list produced. The
first element data_list[[1]] in every DAISIE data list is
the island community metadata, containing the island age and the number
of species in the mainland pool that did not leave descendants on the
island at the present day. This is important information for DAISIE
inference, as it is possible some mainland species colonised the island
but went extinct leaving no trace of their island presence.
data_list[[1]]
#> $island_age
#> [1] 1
#>
#> $not_present
#> [1] 99Next is the first element containing information on island colonists
(every element data_list[[x]] in the list after the
metadata contains information on individual island colonists). This
contains the name of the colonist, the number of missing species, and
the branching times, which is a vector containing the age of the island,
the colonisation time and the times of any cladogenesis events.
Confusingly, it may be that the branching times vector contains no
branching times: when there are only two numbers in the vector these are
the island age followed by the colonisation time. Then there is the
stac, which stands for status of colonist. This is a number which tells
the DAISIE model how to identify the endemicity and colonisation
uncertainty of the island colonist (these
are explained here if you are interested). Lastly, the type1or2
defines which macroevolutionary regime an island colonist is in. By
macroevolutionary regime we mean the set of rates of colonisation,
speciation and extinction for that colonist. Most applications will
assume all island clades have the same regime and thus all are assigned
type 1. However, if there is a priori expectation that
one clade significantly different from the rest, e.g. the Galápagos
finches amongst the other terrestrial birds of the Galápagos archipelago
this clade can be set to type 2.
data_list[[2]]
#> $colonist_name
#> [1] "Plant_g"
#>
#> $branching_times
#> [1] 1.0000000 0.7648553 0.3800341
#>
#> $stac
#> [1] 2
#>
#> $missing_species
#> [1] 0
#>
#> $type1or2
#> [1] 1This data list is now ready to be used in the DAISIE maximum
likelihood inference model from the R package DAISIE. For
more information on the DAISIE data structures and their application in
the DAISIE models see this vignette
on optimising parameters using DAISIE
2. Multiple phylogenies example
In this section we demonstrate an empirical use case of the package on the avifauna of the Galápagos archipelago, which uses several phylogenies for different island colonists.
In the previous example we used a single phylogeny and extracted the colonisation and branching events from it. However, it could be the case that island species have been sampled in different phylogenies (e.g. based on different markers, coming from different studies). Here we look at an example for the terrestrial birds of the Galápagos archipelago. There are 8 time-calibrated phylogenies to extract the colonisation and diversification date from.
First, the phylogenies need to be loaded. The phylogenies are stored
in the DAISIEprep package so we can use the data()
function, the phylogenies could alternatively be loaded using
read.nexus() from the R package ape.
data(coccyzus_tree, package = "DAISIEprep")
data(columbiformes_tree, package = "DAISIEprep")
data(finches_tree, package = "DAISIEprep")
data(mimus_tree, package = "DAISIEprep")
data(myiarchus_tree, package = "DAISIEprep")
data(progne_tree, package = "DAISIEprep")
data(pyrocephalus_tree, package = "DAISIEprep")
data(setophaga_tree, package = "DAISIEprep")Currently the phylogenies are loaded as S3 phylo objects, however, we want to convert them into S4 phylobase objects.
coccyzus_tree <- as(coccyzus_tree, "phylo4")
columbiformes_tree <- as(columbiformes_tree, "phylo4")
finches_tree <- as(finches_tree, "phylo4")
mimus_tree <- as(mimus_tree, "phylo4")
myiarchus_tree <- as(myiarchus_tree, "phylo4")
progne_tree <- as(progne_tree, "phylo4")
pyrocephalus_tree <- as(pyrocephalus_tree, "phylo4")
setophaga_tree <- as(setophaga_tree, "phylo4")Now that all of the phylogenies are loaded we can inspect them. Let’s start with the phylogeny for the genus Coccyzus:
phylobase::plot(coccyzus_tree, cex = 0.1)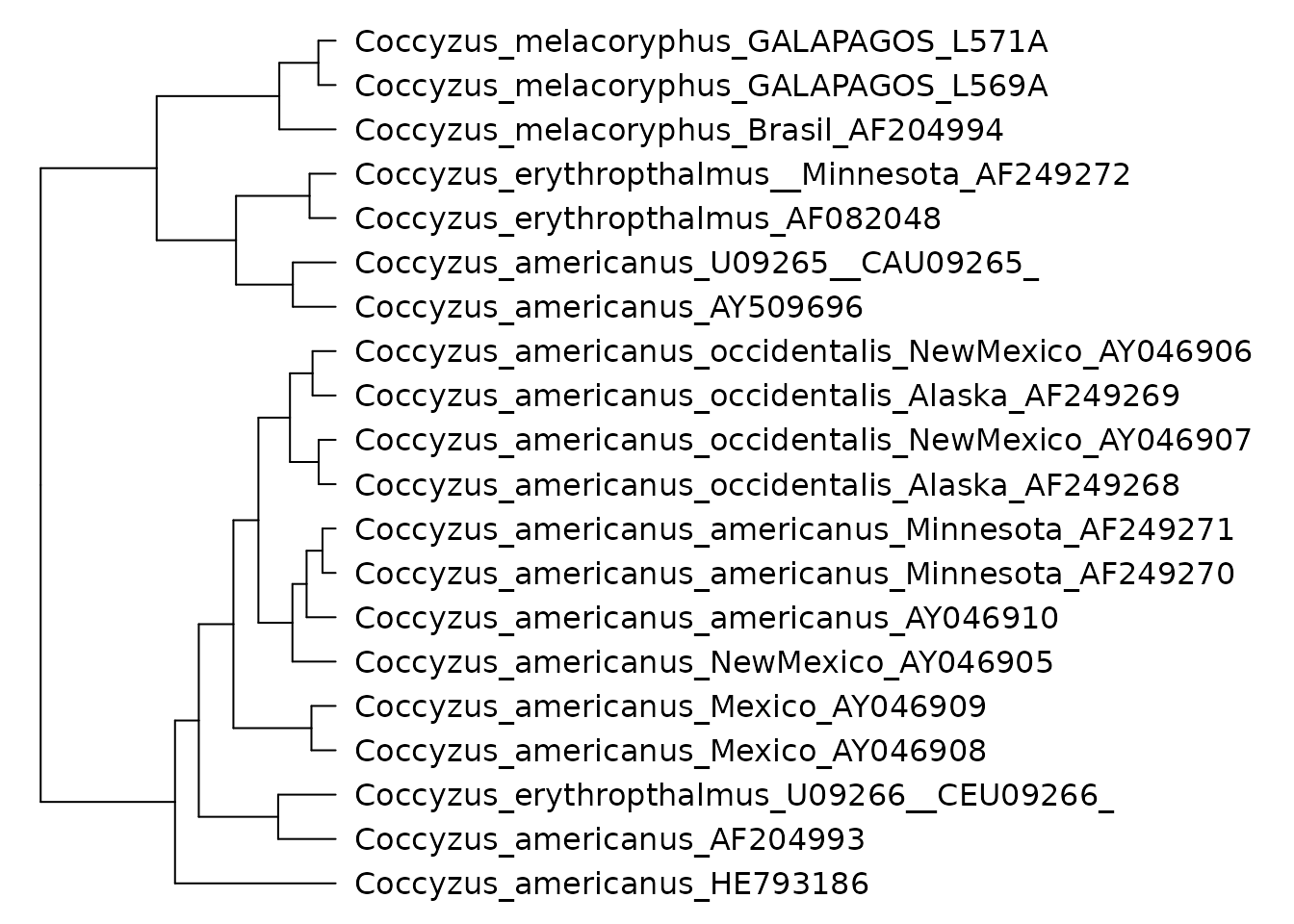
We can now create a table (data frame) of the Coccyzus species that are on the island and their island endemicity status. This table can be imported from a .csv or spreadsheet if you prefer.
The species names on the tree (tips labels) can be extracted using
phylobase::tiplabels(coccyzus_tree). Make sure the
spelling matches exactly including any whitespace and underscores, and
the case of the names.
island_species <- data.frame(
tip_labels = c("Coccyzus_melacoryphus_GALAPAGOS_L569A",
"Coccyzus_melacoryphus_GALAPAGOS_L571A"),
tip_endemicity_status = c("nonendemic", "nonendemic")
)In order to not have to specify the endemicity status for all species
in the phylogeny and instead focus only on the island species, we can
easily assign the endemicity status for the rest of the species in the
tree. Using the island_species data frame produced above,
which specifies the island endemicity status of only the species that
are found on the island, we can generate the rest of the endemicity
statuses for those species that are in the phylogeny but are not present
on the island using create_endemicity_status().
endemicity_status <- create_endemicity_status(
phylo = coccyzus_tree,
island_species = island_species
)Now we have the endemicity status for all Coccyzus species
in the phylogeny, we can combine our phylogenetic data and endemicity
status data into a single data structure, the phylo4d class
from the phylobase R package, in exactly the same way as in
the single phylogeny example.
phylod <- phylobase::phylo4d(coccyzus_tree, endemicity_status)We can visualize the endemicity status of these species on the tree.
plot_phylod(phylod = phylod)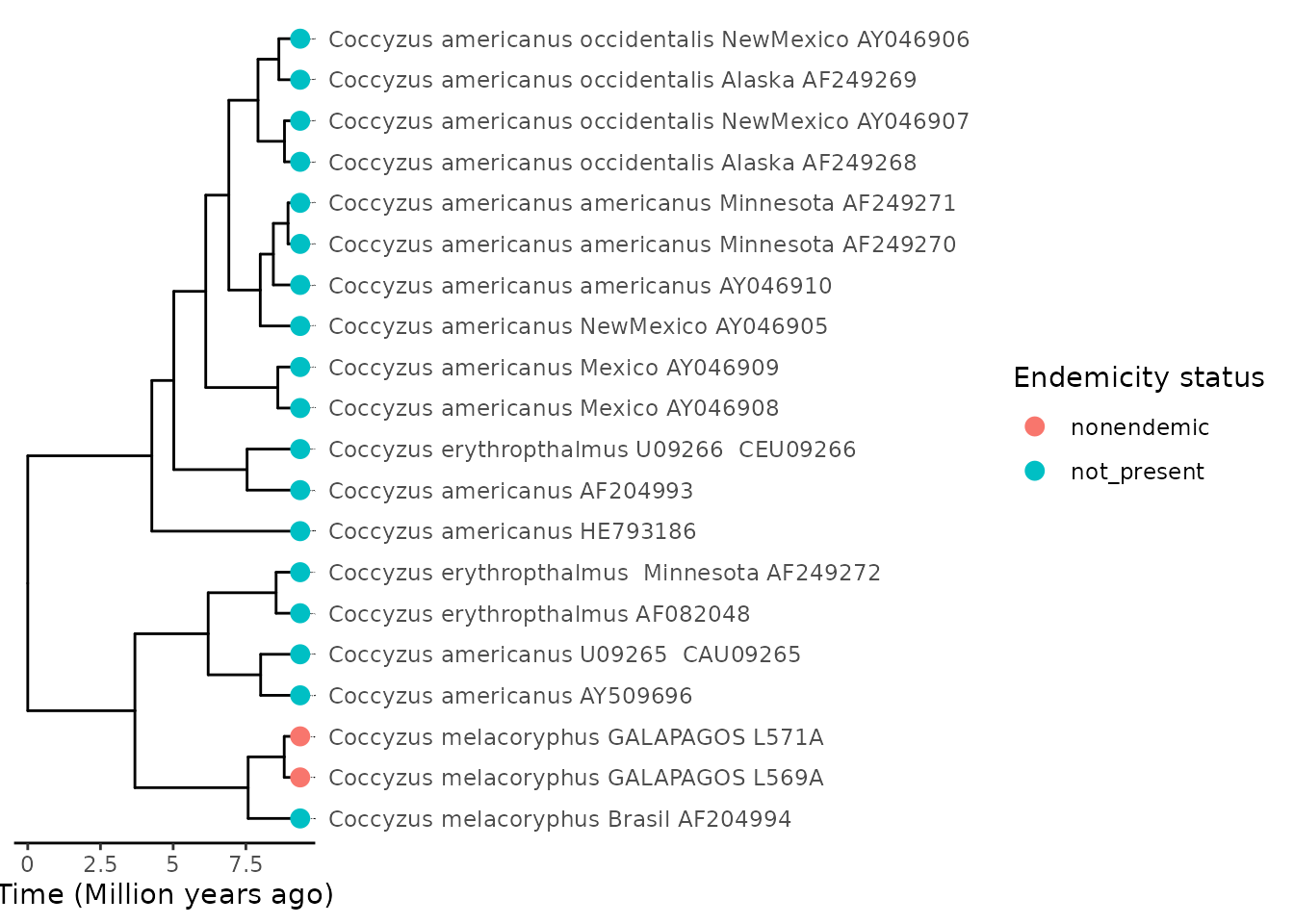
We are now ready to extract the relevant data from the phylogeny, to
produce the island_tbl for the Coccyzus tree. For
this step we use the “asr” method to extract the data which requires
inferring the ancestral geography of each species.
phylod <- add_asr_node_states(
phylod = phylod,
asr_method = "parsimony",
tie_preference = "mainland"
)Plot the phylogeny with the node states:
plot_phylod(phylod = phylod)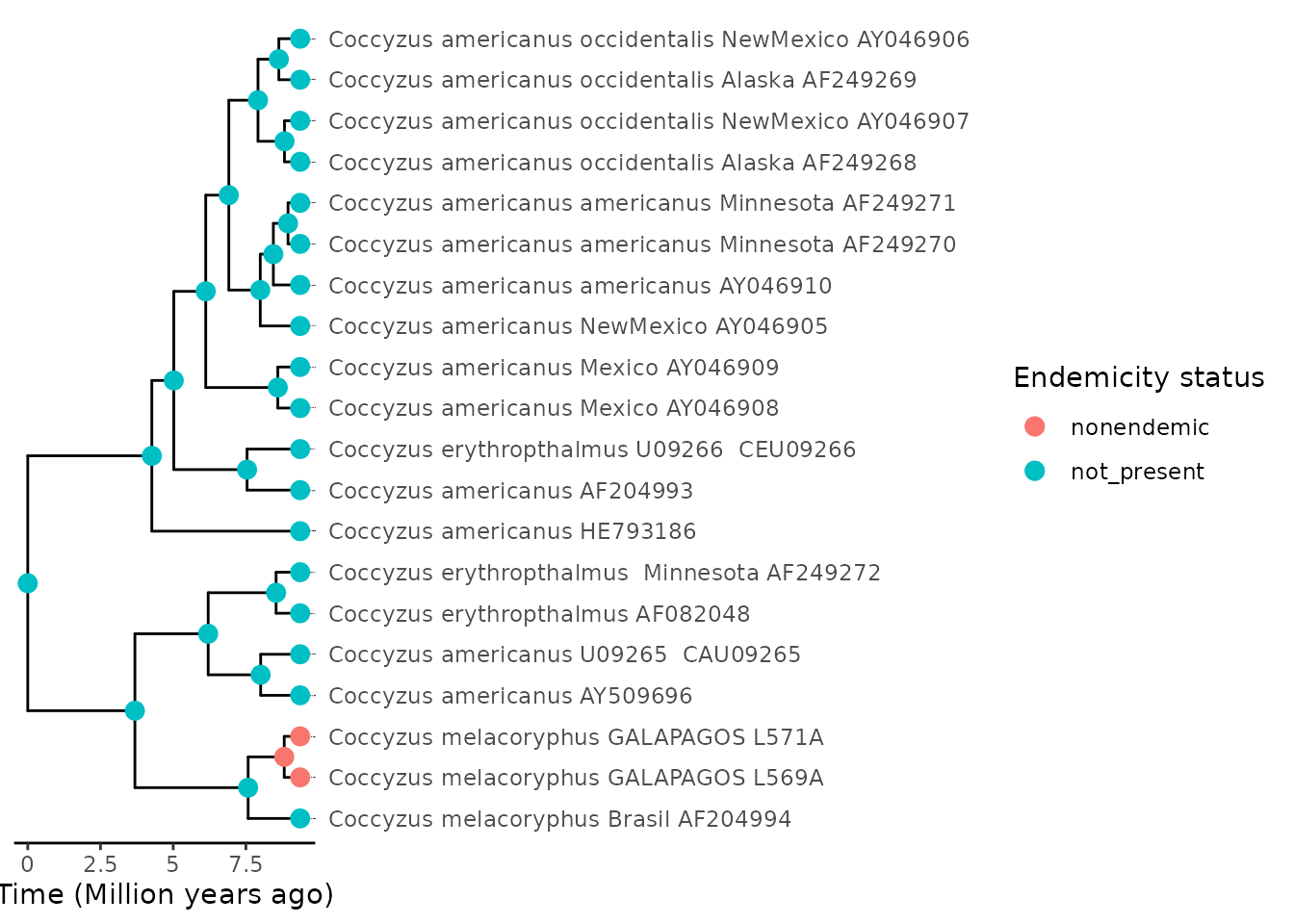
Extract the data from the phylogeny:
island_tbl <- extract_island_species(
phylod = phylod,
extraction_method = "asr"
)
island_tbl
#> Class: Island_tbl
#> clade_name status missing_species col_time col_max_age
#> 1 Coccyzus_melacoryphus nonendemic 0 1.789425 TRUE
#> branching_times min_age species clade_type
#> 1 NA 0.5483906 Coccyzus.... 1Instead of assigning the endemicity to each of the Galapagos bird
phylogenies and converting them to phylo4d objects (as we
did for Coccyzus above ), this has already been done and the
data objects have been prepared in advance and are ready to be used.
data(coccyzus_phylod, package = "DAISIEprep")
data(columbiformes_phylod, package = "DAISIEprep")
data(finches_phylod, package = "DAISIEprep")
data(mimus_phylod, package = "DAISIEprep")
data(myiarchus_phylod, package = "DAISIEprep")
data(progne_phylod, package = "DAISIEprep")
data(pyrocephalus_phylod, package = "DAISIEprep")
data(setophaga_phylod, package = "DAISIEprep")We now have the data for all 8 phylogenies in the correct format,
that is: a dated phylogeny, with tips written in “Genus_species” or
“Genus_species_TAG” format and with the island endemicity status
specified for all tips. We are now ready to extract the island data from
these trees using extract_island_species(), using the “asr”
algorithm.
island_tbl <- extract_island_species(
phylod = coccyzus_phylod,
extraction_method = "asr"
)We can now loop through the rest of the Galapagos phylogenies and add them to the island data.
galapagos_phylod <- list(
coccyzus_phylod, columbiformes_phylod, finches_phylod, mimus_phylod,
myiarchus_phylod, progne_phylod, pyrocephalus_phylod, setophaga_phylod
)
for (phylod in galapagos_phylod) {
island_tbl <- extract_island_species(
phylod = phylod,
extraction_method = "asr",
island_tbl = island_tbl
)
}
#> Warning in extract_species_asr(phylod = phylod, species_label = as.character(phylod@label[i]), : Root of the phylogeny is on the island so the colonisation
#> time from the stem age cannot be collected, colonisation time
#> will be set to infinite.The above example works, but it returns a warning message for the
Darwin’s finches (finches_phylod), because the root state of the
finches’ phylogeny is inferred to be present on the island, as there is
only a single mainland outgroup in the example phylogeny. This means
that the colonisation time will be extracted in asr as
infinite, and then when the island_tbl is converted into a DAISIE data
list this will become a colonist that could have colonised anywhere from
the island origin to the present. For this example this colonisation
time is not a problem, however, for empirical analyses it is recommended
to have many more mainland outgroup species in the tree to ensure the
ancestral state reconstructions can accurately detect the stem age of
the island clade.
plot_phylod(finches_phylod)
island_tbl
#> Class: Island_tbl
#> clade_name status missing_species col_time
#> 1 Coccyzus_melacoryphus nonendemic 0 1.7894251
#> 2 Zenaida_galapagoensis_GALAPAGOS_AF251531 endemic 0 3.1933725
#> 3 C_fus endemic 0 Inf
#> 4 Mimus_macdonaldi_GALAPAGOS_KF411075 endemic 0 4.4853284
#> 5 M_magnirostris_1 endemic 0 0.8544740
#> 6 Progne_modesta_GALAPAGOS_L573A endemic 0 3.0014710
#> 7 Pyrocephalus_dubius_Galapagos_cas01 endemic 0 9.3661766
#> 8 D_petechia_Galapagos_sancris endemic 0 0.1400011
#> col_max_age branching_times min_age species clade_type
#> 1 TRUE NA 0.5483906 Coccyzus.... 1
#> 2 FALSE 0.050253.... NA Zenaida_.... 1
#> 3 FALSE 1.322705.... NA C_fus, C.... 1
#> 4 FALSE 3.680027.... NA Mimus_ma.... 1
#> 5 FALSE 0.222988.... NA M_magnir.... 1
#> 6 FALSE 0.387570.... NA Progne_m.... 1
#> 7 FALSE 0.825248.... NA Pyroceph.... 1
#> 8 FALSE 0.057946.... NA D_petech.... 1Now we have the island_tbl with all the data on the
colonisation, branching times, and composition of each island colonist.
We can convert it to a DAISIE data list to be applied to the DAISIE
inference model. Here we use an island age of the Galápagos archipelago
of 4 million years, and assume that all colonisation time extracted are
precise. Whether they are in fact precise is not covered in this
tutorial, and when using this pipeline to process different data it may
be worth toggling the precise_col_time to
FALSE to check whether assuming uncertainty in colonisation
times influences conclusions.
data_list <- create_daisie_data(
data = island_tbl,
island_age = 4,
num_mainland_species = 100,
precise_col_time = TRUE
)The data_list produced above is now ready for your
DAISIE analyses! See
vignette on optimising parameters using DAISIE
3. Adding missing species
It is often the case that phylogenetic data is not available for some
island species or even entire lineages present in the island community.
But we can still include these species in our DAISIE analyses.
Furthermore, even in the cases where a dated phylogeny does exist, it
may not be open-source and available to use for the extraction. In the
latter cases, it may be possible to know the stem age or crown age if
reported in the literature with the published phylogeny. This section
explains how to use the tools that DAISIEprep provides in
order to handle missing data, and generally to handle species that are
missing and need to be input into the data manually.
For this section, as with the previous section, the core data
structure we are going to work with is the island_tbl. We
will use the island_tbl for the Galápagos birds produced in
the last section.
3.1 Adding missing species to an island clade that has been sampled in the phylogeny
This option is for cases in which a clade has been sampled in the
phylogeny, and at least 1 colonisation or 1 branching time is available,
but 1 or more species were not sampled. For this example, we imagine
that 2 species of Galápagos finch have not been sampled, and that we
want to add them as missing species to the Galápagos finch clade that is
sampled in the phylogeny. The finches have the clade name “C_fus” in the
island_tbl (third row). To assign 2 missing species to this
clade we use following code:
island_tbl <- add_missing_species(
island_tbl = island_tbl,
# num_missing_species equals total species missing
num_missing_species = 2,
# name of a sampled species you want to "add" the missing to
# it can be any in the clade
species_to_add_to = "C_fus"
)The argument species_to_add_to uses a representative
sampled species from that island clade to work out which colonist in the
island_tbl to assign the specified number of missing
species (num_missing_species) to. In this case we used the
species in the clade name, however, this could also have been any
sampled species from the clade, which include:
island_tbl@island_tbl$species[[3]]
#> [1] "C_fus" "C_oliv" "P_cras" "G_diff" "C_pau" "C_par" "C_psi" "C_hel"
#> [9] "C_pal" "G_sep" "G_for" "G_ful" "G_con" "G_mag" "G_scan"With the new missing species added to the island_tbl we
can repeat the conversion steps above using
create_daisie_data() to produce data accepted by the DAISIE
model.
3.2 Adding a lineage with just one species on the island (singleton) when a phylogeny is not available for the lineage, but a colonisation time estimate exists
This option is for adding a singleton lineage (just one
species on the island) when a phylogeny is not available to
conduct the extraction using extract_island_species(), but
an estimate of the stem age of the island colonist is known from the
literature. In this case, we need to input all the information on the
lineage manually ourselves. For illustrative purposes, we use an
imaginary Galápagos bird lineage with 1 species, which is not in our
data set, and fabricate the time of colonisation.
The input needed are:
-
island_tblto add to an existingisland_tbl -
clade_namea name to represent the clade, can either be a specific species from the clade or a genus name, or another name that represent those species -
statuseither “endemic” or “nonendemic” -
missing_speciesIn the case of a lineage with just 1 species (i.e. not an island radiation) the number of missing species to be specified here is zero, as by adding the colonist it already counts as one automatically. -
col_timethe time of colonisation in million years before the present -
col_max_agea boolean (TRUE/FALSE) on whether the colonisation time is precise or should be considered a maximum upper bound on the time of colonisation with some uncertainty -
branching_timesthe times an island clade has speciated in situ on the island. If an island clade has not speciated (i.e. is a singleton) this is NA. (In this it should be NA as the example is for singleton lineages). -
min_ageis the minimum lower bound time of colonisation. This should only be used when the colonisation time added is assumed to be an upper bound (col_max_age=TRUE) -
speciesa vector of species names contained within colonist -
clade_typea number representing which set of rates the colonist is assumed to be under, default is 1, as number greater than one assume this clade is exceptionally different in its colonisation and diversification dynamics
island_tbl <- add_island_colonist(
island_tbl = island_tbl,
clade_name = "Bird_a",
status = "endemic",
# clade with just 1 species, missing_species = 0
# because adding the lineage already counts as 1
missing_species = 0,
col_time = 2.5,
col_max_age = FALSE,
branching_times = NA_real_,
min_age = NA_real_,
species = "Bird_a",
clade_type = 1
)With the new missing species added to the island_tbl we
can repeat the conversion steps above using
create_daisie_data() to produce data accepted by the DAISIE
model.
3.3 Adding a lineage with 2 or more species on the island when a phylogeny is not available for the lineage, but a colonisation time estimate exists
Taking the example above in 3.2, but when the
lineage has 2 or more species. In this case, we we use
an imaginary Galápagos bird lineage with 3 species, which is not in our
data set, and fabricate the time of colonisation.
The input needed are:
-
island_tblto add to an existingisland_tbl -
clade_namea name to represent the clade, can either be a specific species from the clade or a genus name, or another name that represent those species -
statuseither “endemic” or “nonendemic” -
missing_speciesThe number of missing species in this case should ben-1, because adding the lineage manually already counts as 1. -
col_timethe time of colonisation in million years before the present -
col_max_agea boolean (TRUE/FALSE) on whether the colonisation time is precise or should be considered a maximum upper bound on the time of colonisation with some uncertainty -
branching_timesthe times an island clade has speciated in situ on the island. If an island clade has not speciated (i.e. is a singleton) this is NA. -
min_ageis the minimum lower bound time of colonisation. This should be used when an upper bound for colonisation time is known but is different from the crown age. -
speciesa vector of species names contained within colonist -
clade_typea number representing which set of rates the colonist is assumed to be under, default is 1, as number greater than one assume this clade is exceptionally different in its colonisation and diversification dynamics
island_tbl <- add_island_colonist(
island_tbl = island_tbl,
clade_name = "Bird_b",
status = "endemic",
# the total species is 3 and all are missing
# but we add missing_species = 2 because
# adding the lineage already counts as 1
missing_species = 2,
col_time = 2.5,
col_max_age = FALSE,
branching_times = NA_real_,
min_age = NA_real_,
clade_type = 1,
species = c("Bird_b", "Bird_c", "Bird_d")
)With the new missing species added to the island_tbl we
can repeat the conversion steps above using
create_daisie_data() to produce data accepted by the DAISIE
model.
3.4 Adding a lineage when a phylogeny is not available for the lineage, and no colonisation estimate is available.
Taking the examples above in 3.2 and 3.3
but assuming we did not have any phylogenetic data or colonisation time
estimate for the island clade, we could again insert the species as
missing but this time not give the colonisation time. When this colonist
later gets processed by the DAISIE inference model it will be assumed it
colonised the island any time between the island’s formation (in the
case of the Galápagos four million years ago) and the present.
-
missing_speciesIn the case of a lineage with just 1 species (i.e. not an island radiation) the number of missing species is zero, as by adding the colonist it already counts as one automatically. In the case of an island clade of more than one species, the number of missing species in this case should ben-1.
Example for adding lineage with 1 species:
island_tbl <- add_island_colonist(
island_tbl = island_tbl,
clade_name = "Bird_e",
status = "endemic",
# clade with just 1 species, missing_species = 0
# because adding the lineage already counts as 1
missing_species = 0,
col_time = NA_real_,
col_max_age = FALSE,
branching_times = NA_real_,
min_age = NA_real_,
clade_type = 1,
species = "Bird_e"
)Example for adding lineage with 5 species:
island_tbl <- add_island_colonist(
island_tbl = island_tbl,
clade_name = "Bird_f",
status = "endemic",
# the total species is 5 and all are missing
# but we add missing_species = 4 because
# adding the lineage already counts as 1
missing_species = 4,
col_time = NA_real_,
col_max_age = FALSE,
branching_times = NA_real_,
min_age = NA_real_,
clade_type = 1,
species = c("Bird_f", "Bird_g", "Bird_h",
"Bird_i", "Bird_j")
)With the new missing species added to the island_tbl we
can repeat the conversion steps above using
create_daisie_data() to produce data accepted by the DAISIE
model.
3.5 Adding a lineage when a phylogeny is not available for the entire island lineage, but a crown age or minimum colonisation time estimate exists
Taking the example above in 3.2 but assuming we did not
have a colonisation time estimate, but we did have a crown age estimate
or an estimate for the minimum (latest) time the island could have been
colonised by the lineage. When this colonist later gets processed by the
DAISIE inference model it will be assumed it colonised the island any
time between the island’s formation (in the case of the Galápagos four
million years ago) and the crown or minimum age. In the example below we
assume a crown age of 2 million years.
island_tbl <- add_island_colonist(
island_tbl = island_tbl,
clade_name = "Bird_k",
status = "endemic",
missing_species = 0,
col_time = NA_real_,
col_max_age = FALSE,
branching_times = NA_real_,
min_age = 2,
species = "Bird_k",
clade_type = 1
)
#> Warning in add_island_colonist(island_tbl = island_tbl, clade_name = "Bird_k",
#> : Adding a min_age is inconsistent with setting colonisation time to be precise
#> (col_max_age = FALSE). So in this case the min_age is ignored.With the new missing species added to the island_tbl we
can repeat the conversion steps above using
create_daisie_data() to produce data accepted by the DAISIE
model.
data_list <- create_daisie_data(
data = island_tbl,
island_age = 4,
num_mainland_species = 100,
precise_col_time = TRUE
)